

Whatever your personal opinion may be on the COVID-19 pandemic, it has motivated a lot of tech people and companies to use their unique skills and capabilities to fight the virus. This includes the largest supercomputers in the world, such as IBM’s (https://en.wikipedia.org/wiki/Summit_(supercomputer) and the largest data scientists community in the world – Google’s Kaggle https://en.wikipedia.org/wiki/Kaggle. COVID-19 pandemic is already a direct reason for a massive speedup and great advancements in the field of biotechnology.
In this article, you will learn:
- How Summit was able to help scientists working on an effective COVID-19 treatment.
- On a high level – how viruses work and what similarities they have with computer software
- How we can read and later hack the source code of a virus
- To what level can 9000 GPUs optimize a task that would normally take years to compute
- How you can help, with the current I/O problem being worked on by open source developers of AutoDock.
Some context – bear with me please
In an effort to find something that could help us slow down the spread of COVID-19, researchers are turning to super computers, proteins and molecules. But first, let under a bit of the context. Bio-organisms are more like computer programs than you think, and given below is the process of gene transcription that occurs in a lot of bio-systems and all of the cells in your body as you read this article.
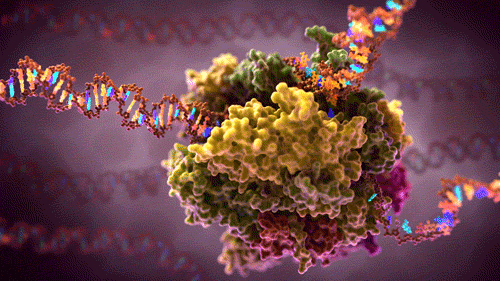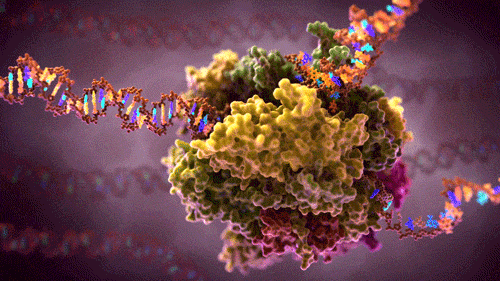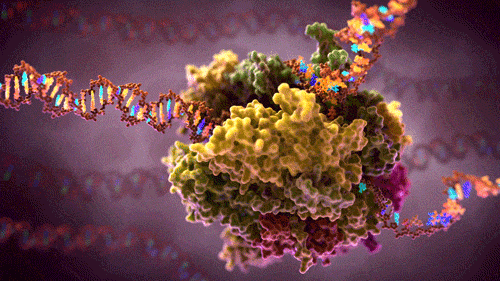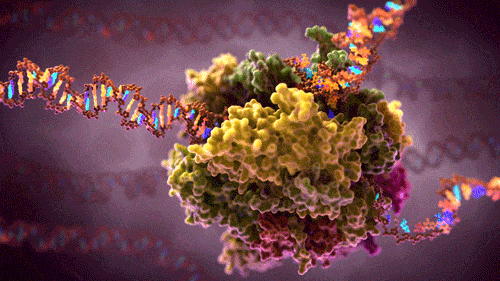
What you see here is a protein complex – many proteins combined together to perform some complex function in a cell .This particular protein-complex floats inside the cell, reads source code (hundreds of megabytes long DNA strains) and converts it into several smaller RNA strains. RNA are instructions carriers, instructions to build other proteins. These free floating RNA instructions will be picked up by builder protein-complexes which will build new proteins depending on the instruction the particular RNA contains. If you like to visualise ideas (like me), here as a video showing and describing this process in 3D.
In software design patterns, our transcribing-protein-complex could be compared to a factory attracting DNA strains to one input and some free floating building blocks to the other input. They then pull the DNA through their inner machinery where one-by-one base pairs of DNA chains are:
- split in half
- analysed
- transcribed to RNA
- glued back together
and the newly created RNA passed to the output and thus creating a new RNA instruction that will float in a cell until some other complex picks it up. The original DNA exits via second output, unmodified.
Hack the genome!

This process happens in most lifeforms and every cell in your body constantly. Viruses have their own protein complexes, allowing them to inject their source code to a human cell. This source code, once inside the cell will be picked up by human factory-proteins and based on its instructions more malicious proteins will be created. There are some steps I didn’t mention here but in a nutshell, this is how virus hijacks human cells. This process is visually explained in this short YouTube video.
There are a lot of protein complexes inside the viruses themselves, each have other capabilities and functions, such as storing energy or working like a suction device, allowing to attach to a human skin. Now this is important for our super computer challenge: what decides about the purpose or capabilities of protein complex is mostly their 3D shape and chemical properties. 3D shape naturally is caused by the combined shape of sub-proteins that build the complex.
The House of Cards
We now have understood that viruses are like small but complex machines or programs and that the 3D shape is an important feature of its protein complexes (sub-components). Let’s see how super computers can help in the fight against COVID-19.
Just like code components, the protein complexes can have some inputs and outputs. These inputs and outputs will actually be physical 3D corridors that formed because of the final shape that protein complex ended up in when all sub proteins linked together. If we want to disrupt the whole virus damaging existence, in theory all we need to do is find some critical protein complexes like the one that allows it to attach to cells and throw a rock in their input or output trying to block it. The problem is finding the right rock that will fit perfectly inside the entry corridor to the protein complex. There are actually several problems here.
First problem is testing, how do we check if the particular rock fits inside a 3D corridor – it’s actually complicated as the rock represents one of the many known and described molecules, and on top of the right shape, it also has to have specific chemical properties that will allow it to glue to the entry corridor and successfully block it.
Second problem is the number of combinations to test. As you can see here, there are over 100 million molecule compounds just in this database. We know their 3D shape so we can model a 3D protein complex with their corridors and try to jam the molecule rocks in them to see if they stick. The thing is each molecule will have to be tested/thrown under different angles. On top of this, protein complexes are not concrete/solid either- they can also bend slightly as they move, thus making a problem space just for a single molecule test fairly complicated.
It’s over 9000!

This video should help you visualise on high level what summit attempted to achieve. As you can probably deduce from previous paragraphs the problem we are facing, while complicated is highly parallelisable – and as we know, GPUs love parallelisation.
What we have here is a problem of modelling 3D shape of molecule and protein and applying some physics on them. Enters… Summit – IBMs super computer. Even with Summit, however, there were some technical challenges. Initially, necessary simulations were calculated to complete in about 4 years on Summit’s 9,216 CPUs, not great, not great at all. Then the attention switched to Summit’s GPUs (27,648 in total). Researchers used OpenCL version of AutoDock, the software used to simulate molecules and proteins binding in 3D. That allowed to transition from CPU to GPU and resulted with a 50x improvement, better, but still not great. Another 3x boost came from rewriting some of the AutoDesk code to use CUDA. Later addition of the OpenMP (an API aiding writing multithreaded, shared memory applications), delivered a final 3x boost. All this allowed for a dataset of 1.4 billion molecules/rocks to be tested against a single protein in about 12 hours! All this work is aimed at creating lists of candidate molecules that can be send to research labs for real live verification.
In case you want to help out
There are still some issues that are being worked on within the AutoDock community, mainly the I/O issues caused by the need of using input files in the computational workflow of finding the right molecules. If you are an opensource enthusiast and want to help, the good people of AutoDock will greatly appreciate anything you can contribute here.
I hope you found this article interesting and as always feel free to leave any of your thoughts in the comments box below.
Author of this blog is Patryk Borowa, Aspire Systems.